Hydrogen Binding Database H2Bind275
Category
Modeling/Simulation
Laboratory
Lawrence Berkeley National Laboratory (LBNL)
Description
Density functional theory (DFT) meets the scaling requirements necessary for binding site characterization in nanoporous frameworks, which gives it a computational and practical advantage for high-throughput screening of potential materials. Since there are numerous types of functionals within DFT with a wide range of analytical and computational complexities, a study of functionals most applicable to hydrogen storage problems is warranted.
To this end, we have identified density functional approximations that provide good performance for hydrogen binding applications with the dataset H2Bind275 that comprehensively represents the hydrogen binding problem capturing the chemical and mechanistic diversity in the binding sites encountered in hydrogen storage materials. Computing reference interaction energies using coupled cluster theory, we have assessed the performance of 55 density functional approximations for predicting H2 interaction energies. Hybrid density functionals (ωB97X-V and ωB97M-V), and double hybrid density functionals (DSD-PBEPBE-D3(BJ) and PBE0-DH), and the semi-local density functional (B97M-V) emerge as the best performing functionals in our study. Based on our results, we recommend using the def2-TZVPP basis set as it represents a good compromise between accuracy and cost, limiting the finite basis set errors to less than 1 kJ/mol.
Status
Online and available for use in collaboration with HyMARC.
Figures
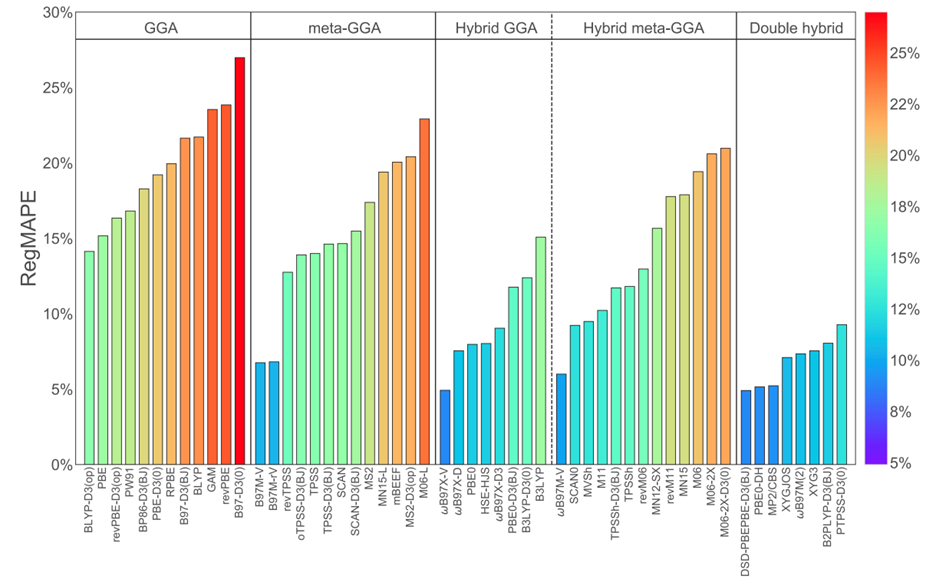
Figure 1. Performance of density functionals for hydrogen storage by rung of Jacob’s ladder
References
- P. Veccham and M. Head-Gordon, J. Chem. Theory Comput. 16 (2020): 4963.